
 返回
返回
常州企业拿下BDD认定背后,藏着中国器械出海的“加速密码”
📌 导语
近日,常州玄言生物科技的“甲状腺乳头状癌术前淋巴结转移检测试剂盒”成功获得美国FDA 突破性医疗器械认定(BDD)。这是常州本土企业首次获此殊荣,消息一出,立刻在医械圈引发热议。
但很多同行看到这条新闻,第一反应都是 ❓问号三连:
拿到BDD就等于拿到FDA上市许可了吗?
BDD到底是个什么级别的“头衔”?
除了BDD,FDA还有没有别的“加速包”?
今天这篇,我们就以常州案例为引,把 FDA医疗器械申报的底层逻辑彻底讲透——
✅ 3大上市路径怎么选
✅ 2大加速通道怎么用
文末还有 2025年中国械企出海的最新数据和趋势,干货满满,建议收藏 📌
2026年5月,玄言生物自主研发的 “甲状腺乳头状癌术前淋巴结转移检测试剂盒(qPCR荧光探针法)” 获得FDA突破性医疗器械认定。
该产品瞄准的是临床一大痛点:甲状腺乳头状癌术前如何精准评估淋巴结转移? 传统方法依赖影像学和术中探查,漏诊率不低。这款产品依托自研的“集成组学+AI”平台,有望在术前给出高精度分子诊断结果,已在上海、江苏、山东等多地开展临床应用。
⚠️ 关键一点:BDD不是上市许可,而是“加速器”。
玄言生物在产品尚未获批上市的阶段就能获得这一认定,说明FDA对其创新价值和临床潜力给予了高度认可。
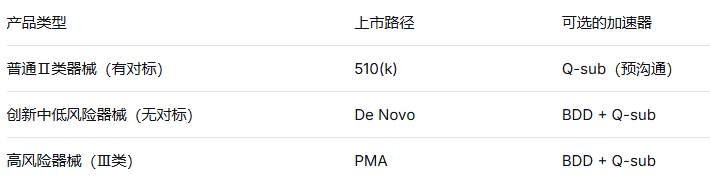
要真正理解BDD的地位,我们得先从FDA的三大核心上市路径说起。
FDA将医疗器械按风险分为Ⅰ、Ⅱ、Ⅲ三类,风险越高监管越严。对应的主要上市路径有三条,搞清楚自己的产品该走哪条路,是申报的第一步。
一句话概括: 找对标、证明“差不多”,是中低风险器械最常走的通道。
✅ 适用对象: Ⅱ类中等风险器械,以及部分不豁免510(k)的Ⅰ类器械。
📊 2025年我国获得FDA认证的医疗器械中,Ⅱ类产品占比高达94.4%,足见510(k)路径的核心地位。
🎯 核心要求: 找到一个已在美国合法上市的“对比器械”(Predicate Device),证明自己的产品在预期用途、技术特性上与其实质性等同。
⏱️ 审评时限: 在MDUFA V框架下,FDA的目标审评周期为 90天。
🧪 适合谁: 普通诊断试剂、监护仪、手术器械等成熟产品,靠性能测试数据即可。
一句话概括: 没有对标产品的中低风险创新器械,通过De Novo开创新分类。
✅ 适用对象: 真正意义上的创新设备,技术全新,找不到合适的对比器械,但风险等级不高(不符合Ⅲ类标准)。
🎯 核心要求: 提交充分的临床证据,证明产品的安全性和有效性,并论证其风险可以通过一般控制或特殊控制得以管理。
⏱️ 审评时限:
法规法定期限: 21 CFR §860.240 规定为 120天
MDUFA V 绩效目标: FDA承诺的审评目标为 150天
(两者不矛盾,法规期限为法定要求,绩效目标为行业承诺)
🧪 适合谁: 新型体外诊断、新型成像设备、新型治疗手段等真正“首创”型产品。
一句话概括: 高风险器械必须走的最严格路径,需要大量临床证据。
✅ 适用对象: Ⅲ类高风险器械(如心脏起搏器、人工心脏瓣膜、植入式神经刺激器等)。
🎯 核心要求: 提交全面充分的科学证据,证明产品安全有效。通常需要完整的临床试验数据、非临床测试报告等。
⏱️ 审评时限: MDUFA V 框架下的目标审评周期为 180天。若涉及专家委员会意见,实际周期通常更长。
🧪 适合谁: 植入式器械、生命支持设备、高风险IVD等,通常需要 3-5年。
路径选对了,不代表只能按部就班地排长队。FDA还为符合条件的创新产品提供了 两条“加速通道” ,帮助企业缩短上市时间。
常州玄言生物走的就是这条路。
BDD是FDA为治疗或诊断危及生命或不可逆衰弱性疾病,且在现有技术上具有显著优势的器械设立的快速通道。它与510(k)、De Novo、PMA等上市路径并行而非替代。
📊 最新数据(截至2025年12月31日):
FDA累计授予1,246项BDD认定
2025财年(2024年10月至2025年9月)授予164项
已有185款获得BDD认定的器械最终获得FDA上市授权
🎁 BDD带来的三大“福利”:
🤝 互动式审评:FDA审评团队与申请人保持密切沟通
⏩ 优先审评:比其他同类产品更快进入审查队列
📄 滚动审评:可分模块提交资料,不用等全部准备好再提交
🎯 哪些产品适合申请BDD?
产品针对严重或危及生命的疾病
现有标准治疗存在显著未被满足的临床需求
器械相比现有手段有显著优势(更精准、更微创、更低风险等)
💡 玄言生物的成功案例恰恰说明:BDD不是等产品上市后才想起申请的“锦上添花”,而应从一开始就纳入产品出海战略布局中。
如果说BDD是“上市阶段的加速器”,那么Q-sub就是 “申报准备阶段的导航仪”。
Q-submission计划是FDA为医疗器械申报者提供的 正式沟通渠道,允许企业在正式提交上市申请前,与FDA讨论产品、提出问题并获得官方反馈。
📅 FDA于 2025年5月 发布了最新版 《Q-Submission Program》最终指南,进一步明确和简化了这一机制。
📌 Q-sub的主要类型:
Pre-sub(预提交): 最常用,提前获得FDA对计划开展的临床研究设计、测试方案、数据收集计划的正式审评与反馈
申报资料问题咨询
研究风险判定
申报资料的信息审查
🎯 Q-sub的核心价值: 企业可以向FDA提问、获得书面或会议反馈,避免因申报方向跑偏而浪费数月甚至一年的时间。咨询问题的精准度直接决定反馈质量,这也是专业法规咨询的核心价值所在。
📊 路径与加速器的组合矩阵
2025年全年,中国医疗器械获得FDA认证总量为 534件,较上年增长4.7%,在连续两年下滑后小幅回升。参与认证的企业数量为 341家,连续三年下滑,行业出清加速。
📌 几个值得关注的核心趋势:
1️⃣ Ⅱ类器械仍是出海主力,占比超94%
2025年获批产品中,Ⅰ类19件、Ⅱ类504件、未分类11件,Ⅱ类占比高达 94.4%。510(k)路径仍是中国械企最主流的出海通道。
2️⃣ 企业数量持续收缩,行业集中度在提升
2022年至2025年,参与FDA认证的国产医疗器械企业数量从410家缩减至341家,参与主体持续精简。
3️⃣ “含金量”在提升
BDD这类高含金量认定的获得不断增加,2025年上半年获批产品结构持续优化。常州玄言生物的BDD认定就是这一趋势的典型例证。
🌏 从地域看,珠三角、长三角企业占据主导,技术驱动型产品占比显著提升。这说明中国械企已不再满足于低附加值的“拿证数量”,而是真正用技术实力在FDA的高门槛上 “拿高分”。
🎯 写在最后
常州玄言生物的BDD认定,是中国医疗器械从“制造”迈向“智造”的一个缩影。
从510(k)到De Novo再到PMA,从Q-sub到BDD——FDA的路径和机制是公平的,但如何选对路、走对方向,考验的是企业的战略眼光和法规专业度。
技术是船,法规是帆。无论是FDA常规注册,还是冲刺BDD这样的“快车道”,专业的法规支持往往是决定成败的关键环节。
📌 关于金飞鹰药械咨询集团
国内领先的医疗器械注册一站式服务机构,自2007年成立以来已服务超5000个二、三类医疗器械项目。提供从产品立项、厂房规划到FDA/CE/MDSAP等多国认证的全流程服务,如需一对一FDA路径评估或BDD申报策略咨询,欢迎联系我们。

