
 返回
返回
📌 核心事实:2026年5月28日,FDA发布了最终版指南《医疗器械上市申请中的人因信息内容》。新指南废除了过去的“优先审查器械清单”,代之以一套基于风险的三级提交分类。所有510(k)、PMA、De Novo、HDE申请均适用。过渡期截止2026年8月1日。
❌ 旧模式:FDA给出一份清单,列明哪些器械类型需要提交人因数据。企业查一下,不在清单上就不管。
✅ 新模式:没有清单了。每个企业自己必须做“使用相关风险分析(URRA)”,根据风险分析结果,判断自己的器械属于三个类别中的哪一个,再按要求提交材料。
⚠️ 后果:过去很多510(k)不需要任何人因数据,现在只要产品有用户交互、且存在“关键任务”,就必须提交完整的人因报告。
🟢 类别1 – 最简版
谁适用:修改后的器械,且修改内容完全不涉及用户界面、用户群体、使用场景、培训或标签。
交什么:一页纸左右的结论和高层摘要。
举个例子:纯后端服务器升级,用户操作界面零变化。
🟡 类别2 – 标准版
谁适用:新器械但没有关键任务;或者修改器械虽改了界面/标签,但不影响任何关键任务。
交什么:用户描述、界面描述、已知使用问题摘要。不需要做总结性验证测试,但必须说清楚为什么没有关键任务。
注意:这种情况比想象中少。大多数与用户直接交互的器械,至少有一个关键任务。
🔴 类别3 – 完整报告
谁适用:新器械存在至少一个关键任务;或修改器械引入/影响了关键任务。
交什么:8个章节的完整人因工程报告,包括URRA、关键任务清单、总结性验证测试数据等。
一句话:绝大多数有用户交互的新器械,默认就是类别3。
FDA的定义很宽:“如果执行错误或未执行,会对患者或用户造成严重伤害的任务。其中,伤害包括医疗救治受损。”
这意味着:漏诊、延误治疗、给错药量——即使没有立即的身体伤害,也可能算“严重伤害”。
⚠️ 一个容易踩坑的技术点:FDA在做使用相关风险分析时,不看错误发生的频率,只看错误可能造成的伤害有多重。这和传统的ISO 14971风险矩阵不一样。
结论摘要(验证测试是否通过)
用户、用途、环境、培训说明
用户界面详解(控件、显示、报警、软件、物理形态、标签…)
已知使用问题(来自同类产品、投诉、文献)
形成性评估摘要(怎么一步步改过来的)
使用相关风险分析(URRA) – 核心章节
关键任务清单 + 认定理由
总结性验证测试详情
✅ 测试人数要求:每个用户群至少15人。两个用户群(如医生+患者)就是30人。
1️⃣ 新增决策点D:即使有关键任务,只要能拿出过硬理由证明现有风险控制已足够,可以不重新做验证测试。这是最大的灵活性提升。
2️⃣ 鼓励用现成数据:以前提交过的内容、类似器械的数据,可以直接引用,不用重复做。
3️⃣ 章节顺序调整:URRA章节提前,报告的逻辑更顺。
4️⃣ 示例大增:附录从无到有,给了三个详细的示例模板(分别对应三类)。还覆盖了儿科用户、AR界面、CGM等复杂场景。
5️⃣ 组合产品单独说明:本指南不完全适用于组合产品,需要联系相应审评中心。
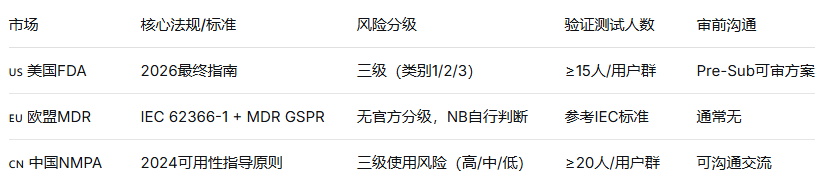
💡 策略建议:如果同时做FDA和欧盟,底层流程用同一套(满足IEC 62366-1即可,再针对FDA调整报告格式)。如果要做NMPA,注意高使用风险器械可能需在中国本地重新做测试。
📱 软件/SaMD/AI器械:人机交互复杂,FDA对AI的人因要求已延伸到“用户是否理解AI的决策逻辑”。
🏠 家用器械:CGM、胰岛素泵、自动注射器等,用户是非专业人员,关键任务多。
⚡ 有源治疗/监护设备:输液泵、呼吸机、监护仪——几乎必属类别3。
💊 组合产品(如预灌封注射器):器械部分仍受本指南约束。
🔄 正在做510(k)修改的企业:原来清关时没要人因数据,现在修改如果影响关键任务,必须补交。
1️⃣ 立刻做URRA:判断你的器械有没有关键任务,落到哪个类别。
2️⃣ 新器械按类别3排期:15人/组的验证测试,从招募到完成至少预留2-3个月。
3️⃣ 把历史人因文档整理好:新指南允许引用以前提交的内容,别浪费。
4️⃣ 尽早做形成性评估:别等到临提交才发现关键任务漏了。
5️⃣ 下载FDA最终指南全文和附录:三个示例模板直接照着写,省时省力。
6️⃣ 关注7月22日的FDA Town Hall:美东时间下午1-2点,问题可提前发至 HFPMET@fda.hhs.gov。
7️⃣ 多市场统一规划:美欧中三套要求底层互通,不要各做各的。
这份新指南的本质信号是:FDA不再满足于你告诉他“器械性能达标”,而是要你用证据证明“用户能安全地用对”。从产品定义的第一天,就把用户研究、任务分析、风险识别纳入开发流程,而不是等到写510(k)文档时才想起人因。
本文基于FDA公开发布的官方指南文件整理,内容仅供参考。

