
 返回
返回如果你正在准备FDA 510(k)或De Novo提交,最近大概率已经被一个消息刷屏了:
eSTAR 6.X将在2026年8月3日正式退役,之后只收7.0。
但真正让人头疼的不是"换个版本"本身。一位器械公司的注册总监在朋友圈里发了一句大实话:"旧模板填了大半年,现在告诉我快过期了?万一赶不上,是要全部推倒重来?"
不一定。但前提是——你得先把下面这六个问题的答案搞清楚。

是死线,但FDA留了一扇窗。
如果你已经在6.X模板中完成了绝大部分内容,并且能在2026年8月3日23:59(美东时间)之前点下"Submit"按钮——恭喜你,不需要做任何版本切换。FDA的接收系统会正常受理你的旧版提交。
同样,如果你收到的是FDA的补充资料通知(AI Response),回函时可以继续使用原始提交所用的模板版本,不受8月3日这个节点影响。
⚠️ 但有一个容易被忽略的陷阱:如果你的提交存在任何不确定性——比如临床试验数据还在收尾、翻译还没完成、内部最终审核还没走完——建议不要再赌旧版了。直接切换到7.0重新整理。因为一旦错过8月3日,旧版将彻底无法使用,届时你面临的是"从零开始",而不是"一键升级"。
💡 一句话建议:能赶就赶,赶不上就别硬赶——果断切7.0反而更省时间。
用一个比喻来说:
eSTAR 7.0 = FDA在过去8个月内所有重大新政的"最终汇合点"。
这不是一次独立的技术更新。从2025年底到2026年夏初,FDA像连发弹夹一样密集抛出了一系列重磅文件:
📅 2025年12月18日 → 真实世界证据(RWE)的指南做了重要修订
📅 2026年1月30日 → 运行多年的检查程序QSIT被全新的CP 7382.850取代
📅 2026年2月2日 → 实施了近30年的质量体系法规QSR正式退场,QMSR上位
📅 2026年5月29日 → 人因工程的终版指南出台
📅 2026年6月1日 → eSTAR 7.0和PreSTAR 3.0同步上线
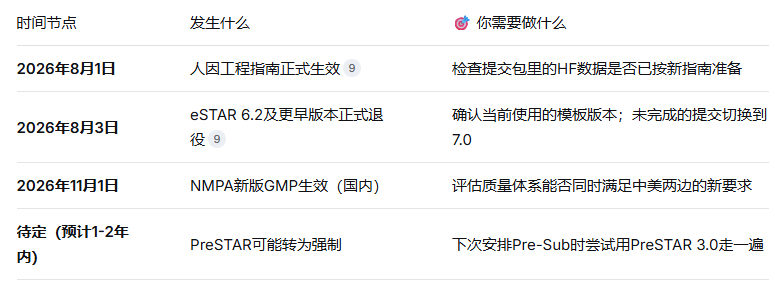
📅 2026年8月1日 → 人因指南正式生效
📅 2026年8月3日 → 旧版eSTAR(6.2及更早)正式退役
这一连串动作不是各自独立的。7.0模板的任务,就是把所有这些新规"收纳"到一个提交工具里。 如果你还在用旧模板,等于绕开了这些新要求——但提交后,FDA审评员依然是按新规来审你的。结果大概率是:你等来的不是批准信,而是一份长长的补充资料通知。
💡 一句话建议:别把7.0看作"升级",把它看作"FDA新规的实体化呈现"。
这是7.0版本中变动最大、也最容易踩坑的部分。
以前人因工程在eSTAR里没有独立模块,很多企业选择"能省则省"。现在不行了。FDA明确表示,eSTAR 7.0已将2026年5月29日发布的人因工程指南内容直接内嵌进了模板。
在"性能测试"板块下,新增了一个专门的区域。你需要在这里完成一个分类判定。怎么判定?看你的产品属于哪种情况:
🟩 如果你的产品是体温计这类操作极其简单的设备 → 系统只要求提交一份简要说明,基本不增加太多负担。
🟨 如果你的产品是输液泵这种需要一定操作培训的设备 → 那就不能只交说明了。你还得额外提交一份使用相关风险分析(URRA),说明你已经识别了哪些操作风险、采取了什么措施来控制。
🟥 如果你的产品是家用呼吸机这类由非专业人员直接操作、操作失误后果可能很严重的高风险设备 → 全套可用性验证测试报告是跑不掉的——包括测试方案、具有用户代表性的受试者数据、测试结论等,一样都不能少。
❓ 实在拿不准自己的产品属于哪一档?系统内置了一个"Guide Me"引导模式,会通过一系列问题帮你逐步定位到合适的类别。
选错了会有什么后果?
🔻 选低了(本该报高风险类别却报了低风险)→ FDA审评时发现人因数据不足,直接发AI(补充资料通知),答复周期至少90天起跳。
🔺 选高了(只需要低风险类别却报了高风险)→ 白花了大量时间和预算做了一堆不必要的测试。
💡 一句话建议:拿不准就选"Guide Me"让系统帮你判断。另外注意一个关键时间点——人因指南8月1日正式生效,比模板退役(8月3日)还早两天。这意味着你提交时,FDA已经按新指南的标准在审了。
2026年2月2日,FDA的QMSR(质量管理体系法规) 正式取代了实施近30年的QSR(21 CFR Part 820) 。
这不是换了个缩写这么简单。实质是FDA把ISO 13485:2016以引用方式纳入了美国法律框架。
以前,FDA的质量体系要求(QSR 820)和ISO 13485是两套独立的标准。企业在准备文档时往往要做一张"对照表"——这边对应FDA的820条款,那边对应ISO的条款。现在,两者的底层框架统一了。
⚠️ 但不要想当然地认为"完全一样"。QMSR在ISO 13485的基础上,增加了FDA独有的一些细化要求,比如:
对供应商管理的某些具体规定,比ISO 13485更细
投诉处理(Complaint Handling)需要遵循FDA特有的流程和时限
纠正预防措施(CAPA)的记录和报告格式,FDA有明确要求
在eSTAR 7.0里,质量体系相关的填写字段已经按照QMSR/ISO 13485的逻辑重新排列过了。如果你的内部质量文档还是按老QSR条款编号整理的,在新模板里你会遇到一个尴尬的情况——"找不到对应的格子填"。
📌 另外:FDA明确表示不会要求提供ISO 13485认证证书。
💡 一句话建议:不需要为了FDA专门去做ISO 13485认证,但你的质量体系必须能满足ISO 13485的全部要求。审评员不看你的证书,但会看你在eSTAR里填的内容是不是跟ISO 13485对得上。
非常有用。这可能是7.0里对你最有利的一个变化。
2025年12月18日,FDA更新了《使用真实世界证据支持医疗器械监管决策》指南,取代了2017年的旧版指南。
里面有一个突破性的调整:
在特定条件下,FDA不再强制要求提交可识别个人身份的患者级数据,允许使用去标识化的大型数据库——比如器械注册登记数据、医保理赔数据库、电子健康档案等。
eSTAR 7.0的RWE模块已经按照这份最新指南重新设计了。它专门为"去标识化数据"开辟了独立的上传路径,并用结构化的填写字段引导你说明:
🔹 数据是从哪里来的?
🔹 去标识化是怎么操作的?(是否符合HIPAA的安全港标准?)
🔹 数据的质量怎么保证?
🔹 为什么这些数据足以证明产品的安全性和有效性?
谁最应该关注这个变化?
👥 产品已经在市场上用了好几年、积累了大量的真实使用数据
👥 产品适应症比较广,做RCT成本太高或伦理上通不过
👥 做的是罕见病相关器械,患者数量少到根本撑不起大规模RCT
⚠️ 不过要泼一盆冷水:RWE不是"免临床"的代名词。FDA对数据质量和相关性的要求依然严格。不是随便从哪个数据库导出一张表就能用的。eSTAR 7.0里那些结构化的填写字段,本质上就是在倒逼你论证"为什么你的数据值得被采信"——这本身就是一个需要认真准备的工程。
💡 一句话建议:如果RWE是你在考虑的路径,现在就可以开始梳理数据库资源了——确认你的数据源能否导出符合FDA去标识化要求的格式。
PreSTAR是eSTAR的"前传"模板,用来准备上市前跟FDA的各种沟通会议。
以前你可能安排过Pre-Sub(预提交会议),那时候提交的资料格式是自由的——自己做一份PPT或PDF就发出去了。现在FDA希望这个过程也标准化,走PreSTAR模板。
PreSTAR 3.0最大的变化是适用范围大幅扩大了。
之前的2.X版本只支持三类:Pre-Submission(预提交会议)、IDE(研究性器械豁免)和513(g)分类请求。
3.0新增支持了以下场景:
📎 Submission Issue Requests(提交问题请求)
📎 Informational Meetings(信息性会议)
📎 Study Risk Determinations(研究风险判定)
📎 PMA Day 100 Meetings(PMA第100天关键节点会议)
📎 Accessory Classification Requests(附件分类请求)
基本上你能想到的需要跟FDA坐下来聊的情形,它都覆盖了。
⚠️ 虽然目前PreSTAR还是自愿使用,但以FDA的惯常操作来看——510(k)和De Novo都是先推自愿、再过渡到强制——PreSTAR的"转正"只是时间问题。相关的草案指南已经发布了,终版出来之后一年内大概率落地。
💡 一句话建议:如果你是第一次向FDA提交,或者产品的分类归属不太清晰,强烈建议在正式提交之前安排一次Pre-Sub。用PreSTAR 3.0准备会议材料,把产品分类、谓词选择、临床策略这些关键问题提前跟FDA确认清楚。这比正式提交后被"打回来"重新折腾要高效太多了。
把上面六个问题的答案串在一起,你会看到一个清晰的趋势:
FDA正在把"法规条文"翻译成"模板里的必填字段"。
过去,经验丰富的RA专员可以通过专业判断来决定"哪些数据放进去、哪些可以不放"。现在eSTAR替你做了这个判断——所有问题都预设好了,漏任何一个都直接锁死"提交"按钮。
落实到行动层面,几个明确的信号:
🧩 人因工程不能再拖到提交前"补作业",要从产品设计阶段就开始规划
📋 质量体系文档不能只按老FDA条款整理,需要向ISO 13485的框架靠拢
📊 临床策略如果打算走RWE路径,要提前确认数据源是否符合去标识化要求
⏰ 项目排期里至少要给"适应新模板"留出2-4周的缓冲时间
🤝 Pre-Sub沟通建议前置,用PreSTAR 3.0先跟FDA对齐预期
一句话收尾:eSTAR 7.0不是一次"版本更新",它是FDA监管逻辑的升级——模板即法规。把这个逻辑理解透的人,把规则变成工具;被动等着"被通知"的人,只会把规则变成门槛。
本文信息截至2026年6月,基于FDA官方发布的eSTAR 7.0、PreSTAR 3.0及相关指南文件整理,具体策略请以FDA最新公告为准。

