
 返回
返回上一期我们了解了UDI是什么。这一期,我们来解决一个更实际的问题:作为医疗器械生产企业,UDI合规到底要做什么?用一句话概括——创建编码、赋予载体、上传数据。听起来简单,但每一步都有严格的技术要求和法规门槛。

根据2019年8月国家药监局发布的《医疗器械唯一标识系统规则》(2019年第66号公告,以下简称《规则》),医疗器械唯一标识系统由医疗器械唯一标识(UDI)、数据载体和数据库三部分组成。对应到企业端的实际操作,就是创建编码、赋码、数据上传三件核心工作。
《规则》明确要求:相关医疗器械产品应当在生产过程中赋予医疗器械唯一标识,在产品上市销售前应当完成医疗器械唯一标识产品标识和相关数据的上传。三件事环环相扣,缺一不可。
这是UDI合规的起点。企业需要按照所选择的发码机构的编码规则,为医疗器械产品创建唯一标识。
📌 Step 1:确定实施UDI的产品范围
首先要明确哪些产品需要实施UDI。根据三部门2026年第21号公告:
🔴 2027年6月1日起:全部第二类医疗器械(含体外诊断试剂)+ 全部第一类体外诊断试剂
🔴 2029年6月1日起:全部第一类医疗器械
如果产品属于2026年第15号公告明确的七大类豁免情形,可免于开展UDI创建、赋码和数据库提交。不属于豁免范围的产品,必须依法实施。
📌 Step 2:选定发码机构,申请厂商识别代码
在中国,企业可以选择国家药监局认可的发码机构。目前国家药监局UDI数据库中列出的发码机构主要有:
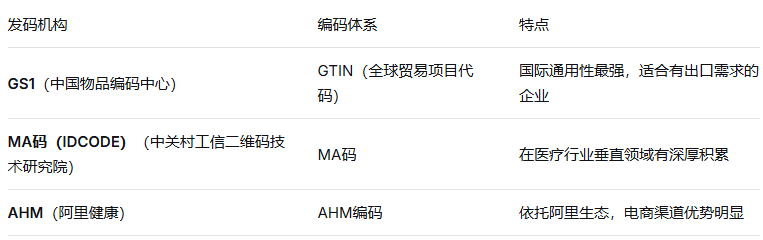
⚠️ 特别提醒:一旦选定发码机构,DI的编码规则就确定了,中途更换成本极高——涉及重新编码、重新赋码、重新上传数据库等一系列工作。企业应结合自身产品类型、国内外目标市场、销售渠道等因素慎重选择。
📌 Step 3:分配产品标识(UDI-DI)
UDI-DI是UDI的静态部分,是识别注册人/备案人、医疗器械型号规格和包装的唯一代码。
根据《规则》及YY/T 1879-2022《医疗器械唯一标识的创建和赋予》,分配DI需遵循以下原则:
✅ 稳定性原则:当医疗器械的基本特征未发生变化时,UDI-DI应保持不变
⚠️ 变更原则:如果出现可能导致医疗器械错误识别或可追溯性不明确的变化(如包装中产品数量变化、无菌包装属性变化、产品型号/规格变化等),必须分配新的UDI-DI
📦 包装级别原则:应为医疗器械的最小销售单元分配UDI,更高级别的包装(不含运输包装)应具有各自的UDI
📦 使用单元原则:当最小销售单元中包含多个相同的使用单元时,应为使用单元分配UoU UDI-DI并存储在UDI数据库中
📌 Step 4:分配生产标识(UDI-PI)
UDI-PI是UDI的动态部分,包括与生产过程相关的信息:产品批号、序列号、生产日期、失效日期等。PI的组成宜采用与标签内容保持一致的原则——标签上包含哪些信息,PI中就包含哪些信息。
在GS1标准中,常用应用标识符(AI)如下:
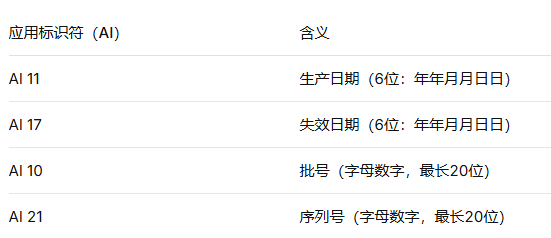
⚠️ 关键提示:UDI-PI的日期格式应严格满足发码机构的编码规则要求——根据GS1标准,UDI载体日期格式为6位数字(YYMMDD,即年年月月日日) ,企业应注意与日常习惯的日期格式区分。
编码创建完成后,需要将UDI以可识读的方式呈现在产品上——这就是“赋码”。
📌 Step 1:确定UDI数据载体
UDI数据载体是存储或传输医疗器械唯一标识的数据媒介。常见的数据载体包括:
📊 一维码(如GS1-128):技术成熟,成本低,但信息容量有限
📱 二维码(如GS1 DataMatrix):信息容量大,占用空间小,纠错能力强
📡 RFID射频标签:可非接触式读取,适用于高值器械
企业需要根据产品特征、包装空间、应用场景等因素综合选择合适的数据载体。
📌 Step 2:确保人工可识读部分(HRI)
根据《规则》,UDI数据载体应当同时包含人工可识读部分(HRI,Human Readable Interpretation) ——即用数字和字母直接呈现的UDI编码,方便人眼识别和核对。
📌 Step 3:在产品或包装上赋码
赋码的具体位置和方式因产品而异:
📦 对于有包装的医疗器械,通常赋在最小销售单元包装上
🔧 对于重复使用器械,应采用本体直接标记(DPM,Direct Part Marking)
💻 对于独立软件(非物理介质提供),无需实体载体,应在用户界面以易读的纯文本格式显示UDI
⚠️ 关键提示:不同包装级别应有各自的UDI-DI。最小销售单元、更高级别包装(不含运输包装)均应赋码。
编码创建了,标签也贴上了,但工作还没有结束。未完成数据库上传的UDI,等同于“假码” 。
📌 Step 1:登录系统
企业通过国家药品监督管理局网上办事大厅(https://zwfw.nmpa.gov.cn/)进行法人登录,进入医疗器械唯一标识管理信息系统。
📌 Step 2:选择申报方式并提交数据
国家药监局UDI数据库为注册人/备案人提供三种数据申报途径:

📌 Step 3:确保数据准确并与医保信息关联
根据2026年第21号公告的要求:
📅 相应实施日期起生产的医疗器械,在上市销售前,注册人、备案人应按照相关标准或规范要求,将最小销售单元、更高级别包装的产品标识和相关数据上传至医疗器械唯一标识数据库
✅ 确保数据真实、准确、完整、可追溯
🔄 当医疗器械最小销售单元产品标识相关数据发生变化时,应当在产品上市销售前在数据库中进行变更
➕ 医疗器械最小销售单元产品标识变化时,应当按照新增产品标识在数据库中上传数据
🏥 对于已在国家医保局医保医用耗材、医保体外诊断试剂等医保分类与代码数据库中维护信息的医疗器械,要在UDI数据库中补充完善医保分类与代码字段,同时在医保数据库中维护UDI相关信息,并确认两者数据的一致性
⚠️ 关键提示:未在UDI数据库中完成上传的UDI,等于没有UDI。未完成UDI赋码、未在国家UDI数据库上传数据、未完成医保编码映射关联的产品,医疗机构无法扫码入库、医保系统不予结算报销——直接影响到产品的市场准入和销售。
让我们重新梳理一下UDI合规的完整链条:
第一件事:创建编码
确定产品范围 → 选定发码机构 → 申请厂商识别代码 → 分配UDI-DI → 分配UDI-PI
↓
第二件事:赋码
选择数据载体 → 设计标签(含HRI) → 在产品/包装上赋予UDI
↓
第三件事:数据上传
登录系统 → 选择申报方式 → 提交UDI数据 → 关联医保信息 → 持续维护更新
三件事,环环相扣,缺一不可。
《医疗器械生产监督管理办法》明确要求医疗器械注册人、备案人、受托生产企业应当按照国家实施医疗器械唯一标识的有关要求,开展赋码、数据上传和维护更新,保证信息真实、准确、完整和可追溯。
与此同时,新版《医疗器械生产质量管理规范》(国家药监局2025年第107号公告)已于2026年11月1日正式施行,进一步将UDI合规纳入生产质量管理体系的核心要求。
2027年6月1日的倒计时已经开始,这场覆盖全行业的“质量溯源革命”没有旁观者。
从编码创建到标签打印,从数据维护到官方数据库对接——每一步都有技术门槛和合规要求。选择一套专业、可靠的UDI管理工具,能让企业少走弯路、事半功倍。
⚙️ 金飞鹰UDI管理软件,专为医疗药械生产企业量身打造,覆盖UDI编码生成 → 标签打印 → 数据维护 → 官方数据库对接全流程。无论您需要对接中国NMPA-UDI数据库、美国FDA GUDID还是欧盟EUDAMED,金飞鹰都能为您提供一站式解决方案,助力企业轻松完成产品全球上市申报。
✅ 支持GS1、MA、AHM等多种发码机构编码标准
✅ 自定义标签模板,一维码、二维码、RFID随心选择
✅ 三种数据申报方式灵活可选,与ERP/OA等主流系统无缝对接
✅ 一次配置,全球申报,让跨境合规一步到位
📌 下一期预告: UDI实施时间线全面梳理——你的产品还剩多少时间?我们将详细解读三类、二类、一类医疗器械的UDI实施时间节点,帮你算清楚合规“倒计时”。敬请期待!
📱 咨询热线:车金金 18628333580
📍 公司总部:深圳市南山区前海路3101号振业国际商务中心2405
📍 广州分公司:广州市黄埔区科学大道50号绿地中央广场A3栋1905-1907
⏳ 合规不等人,立即行动!

